Our group is interested in rare and undiagnosed immunological diseases that mainly present as autoimmunity, severe allergy, infection susceptibility, and early cancer. Some of the diseases are caused by yet unknown infectious agents, clonal genetic mutations acquired later in life, or other environmental factors. Others are caused by hereditary single gene defects in pathways important for immune cell function.
In hereditary immune diseases, a complex interplay between environment and other genetic variants exist, and leads to a broad array of clinical diseases. The penetrance is often incomplete, meaning that one family member can get a very severe disease, whereas another stays asymptomatic. The same mutation can also lead to seemingly opposed phenotypes. One patient can develop a life-threatening, multisystemic autoimmune disease, whereas another with the same mutation might suffer from recurrent infections.
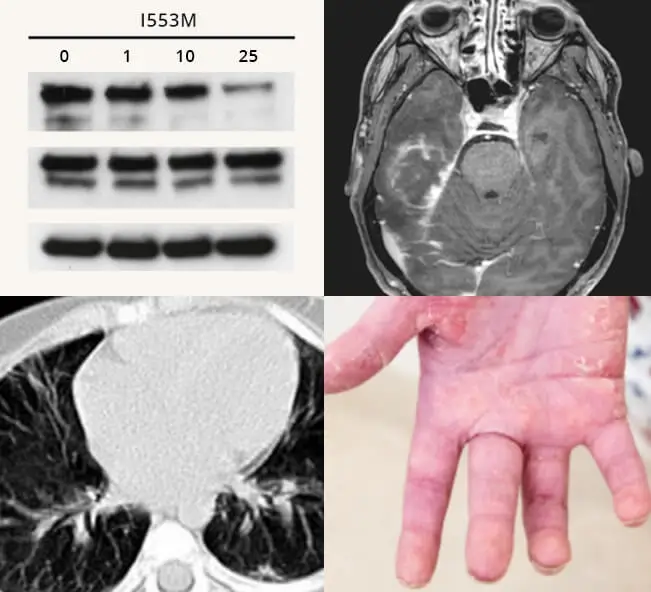
Example presentations of rare immune diseases. Upper left: western blots of patient lymphocytes harboring a NFKB1 mutation; increased concentrations of TNF lead to the loss of the full-length protein product (see publication). Upper right: Ebstein-Barr virus (EBV) -driven central nervous system lymphoma in an adult patient with Wiskott-Aldrich syndrome. Lower left: Lung disease in an eight-year-old girl. Lower right: Psoriasis in a six-year-old boy.
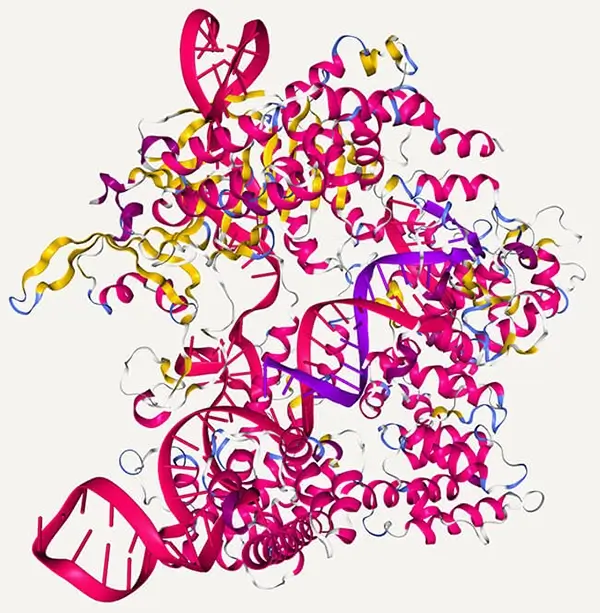
Crystal structure of Cas9 in complex with guide RNA and target DNA. The structure is originally published in Nishimasu, H. et al. Cell 2014.
Genome editing in hereditary immune diseases
CRISPR-Cas9 is a new gene editing technology that can cut genome in designed locations. In patients with genetic diseases, CRISPR-Cas9 can cut the disease-causing mutation, followed by the cell repairing the cut site to a normal sequence, provided that the cell has access to a repair DNA. While the cutting is efficient, the repair that follows is inefficient, and usually silences the gene. The unmodified CRISPR system can also cause large genomic rearrangements and alter the DNA damage response, thus predisposing cells to cancer.
Our goal is to improve the precision mutation correction by altering the pathways that govern DNA repair. We have attached DNA repair proteins to Cas9 to bias editing locally, timed the editing with cell cycle. Our long-term goal is to develop a platform that can take any patient mutation and correct it to a “healthy” sequence, thus treating a large number of genetic diseases. Besides gene therapy, the platform could be adapted for other applications such as cell editing for cancer immunotherapy.
Personalised use of targeted therapies in rare diseases
Many genetic immune diseases harbor inherited mutations in pathways that can be targeted with novel therapies originally developed for cancer and autoimmune diseases. As an example, JAK inhibitor Ruxolitinib can target activating STAT3 mutations that cause early-onset multi-organ autoimmune disease. Compared to cell therapies and gene editing, the use of targeted drugs is a much faster and safer way to improve patient symptoms and quality of life.
For mutant genes that localize to known immunological pathways, the choice of targeted drugs is often easy. However, in several mutations, the function of the gene is poorly known. One of our future projects is to systematically catalogue poorly characterized mutant protein interactomes in immune cells, and use the information to rationally choose drugs for diseases in which no obvious druggable targets exist.
In many patients with suspected genetic immunological disease, the mutation that causes their disease is either undruggable or not found despite extensive genome-wide sequencing studies. Our second future goal is to utilise transcriptome profiling to detect immunological profiles that the new therapies can target, and tailor the treatment based on the sequencing result.
This would enable the rational use of biologics and small molecule inhibitors in patients who either lack a diagnosis or have a genetic mutation that can’t be directly targeted with existing therapies.
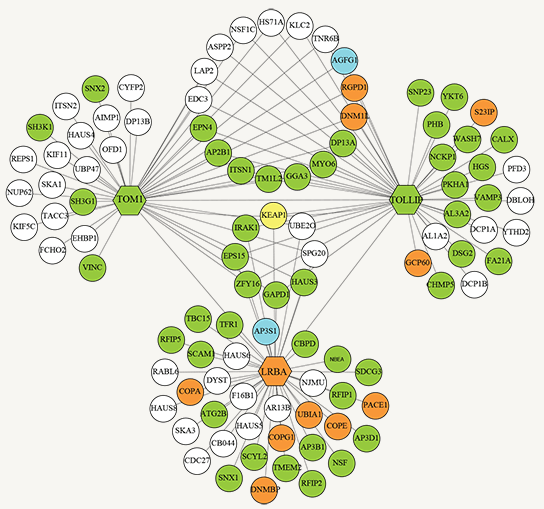
Protein interactomes can highlight connections between mutant proteins and signaling pathways that can be targeted with existing drugs. The figure shows interactomes of TOM1 and LRBA proteins, both of which function in the endolysosomal compartment and cause multi-organ autoimmune disease when mutated.
This article (in Norwegian) from Bioteknologirådet explores the global impact of Crispr technology, including its ongoing patent disputes and medical advancements. It features our group leader, Emma, who discusses our work at the University of Oslo to develop personalised Crispr-based treatments for rare immunodeficiency diseases.
This Swedish article from Barncancerfonden highlights new research on the genetic factors behind childhood cancer. Emma discusses the role of genetic screening in identifying inherited cancer risks and how this knowledge can improve patient care.
This Norwegian article from Apollon explores the potential of CRISPR in medicine. Emma discusses how our research aims to develop safer and more precise gene-editing treatments for immunodeficiency diseases.
We are on Addgene!
Our lab has deposited materials at Addgene for distribution to the research community. Addgene is a nonprofit plasmid repository dedicated to improving life science research.