Home » Publications
Pavel Kopcil, Carolina W. Ervik, Ganna Reint, Katariina Mamia, Monika Szymanska, Shiva Dahal-Koirala, Jacob Conradi, Sigrid Fu Skjelbostad, Oda A. Dønåsen, Xiaojun Jiang, Cecilia Fahlquist-Hagert, Oddrun Kristiansen, Trond M. Michelsen, Espen Melum, Rasmus O. Bak, Anna Komisarczuk, Emma Haapaniemi
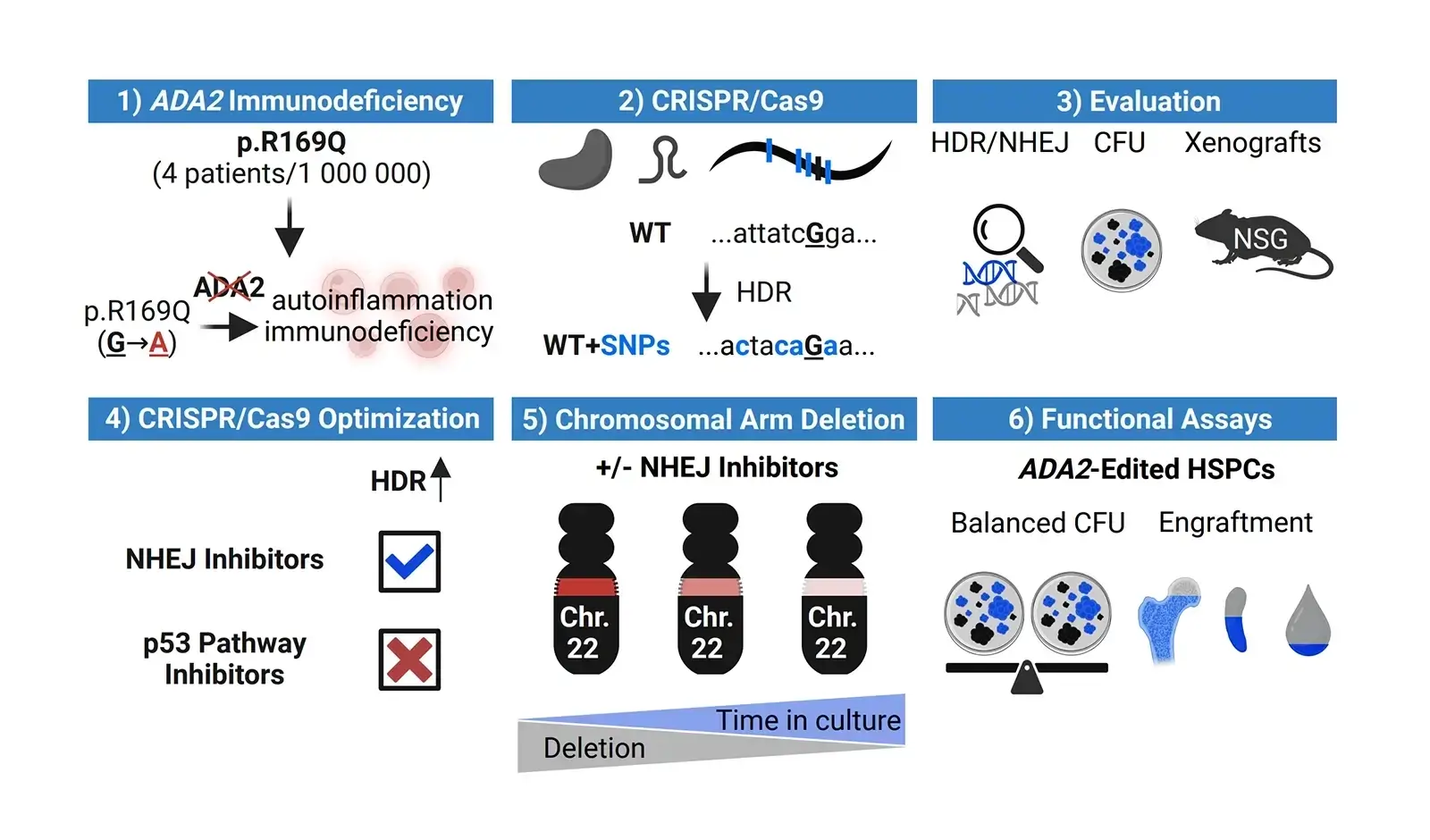
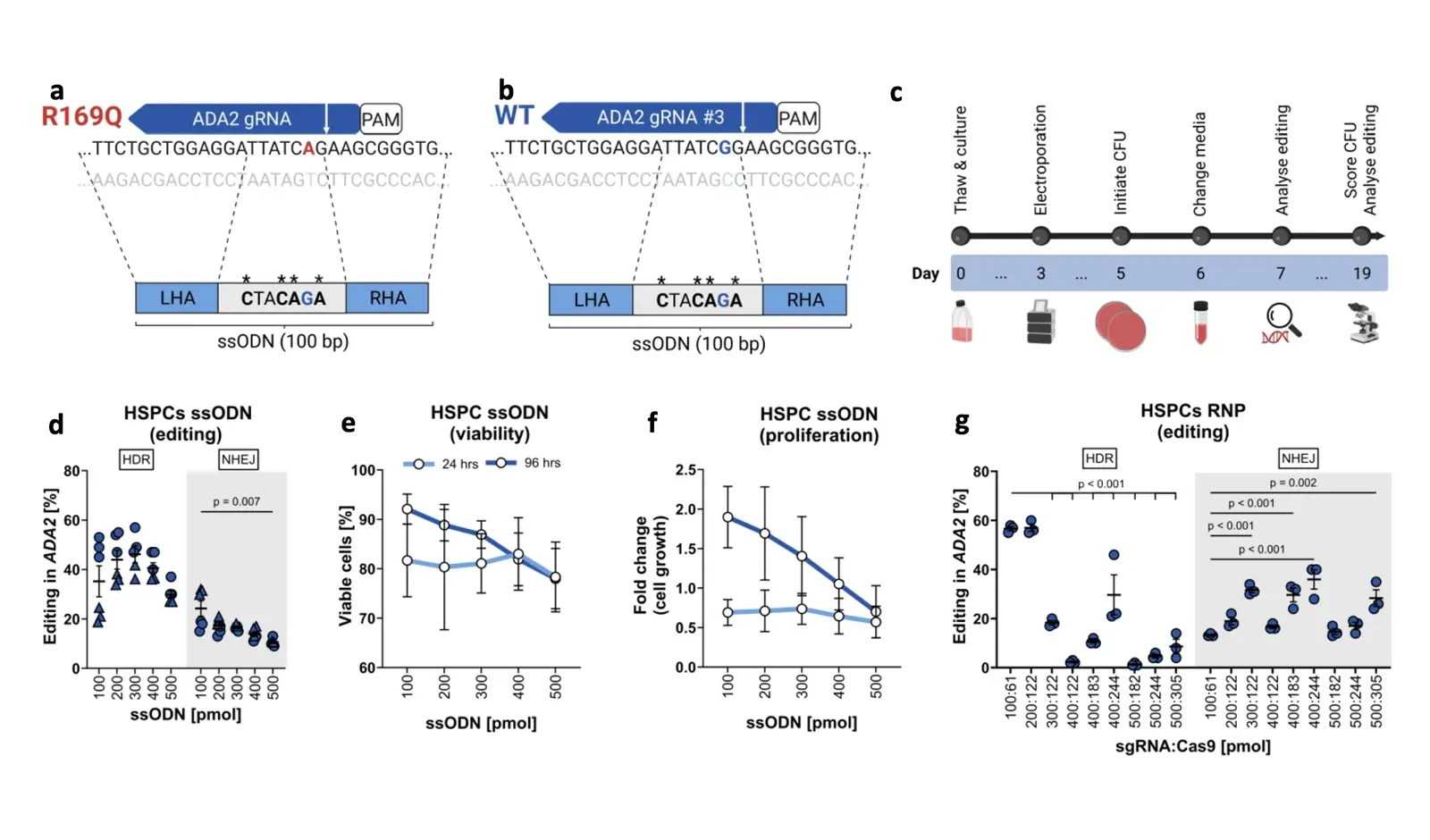
Background: The homozygous ADA2: c.506G>A (p.Arg169Gln; p.R169Q) variant accounts for majority of Deficiency in Adenosine Deaminase 2 (DADA2). This monogenic disorder may be amenable to ex vivo gene therapy by correcting the pathogenic mutation in CD34+ hematopoietic stem and progenitor cells (HSPCs).
Objective: To apply CRISPR-Cas9 and homology-directed repair (HDR) as a surrogate strategy to model correction of the pathogenic ADA2 c.506G>A variant in healthy cord blood HSPCs.
Methods: HSPCs were electroporated with optimised CRISPR-Cas9 editing reagents, and editing outcomes, including HDR and on-target deletions, were quantified by ddPCR. Cell functionality was assessed through colony-forming unit (CFU) assays and by xenotransplantation into NOD SCID Gamma (NSG) mice. Two HDR enhancement strategies were tested: (1) genetic inhibitors of p53 and non-homologous end joining (NHEJ) pathways, and (2) pharmacological NHEJ inhibition.
Results: Small-molecule NHEJ inhibitors increased HDR efficiency approximately two-fold (from ∼40 % to ∼80 %). Edited HSPCs retained normal CFU capacity and successfully engrafted in NSG mice. However, up to 8 % of edited cells exhibited on-target chromosome loss, though this declined over time. Up to 40 % of T cells and fibroblasts demonstrated similar losses under NHEJ inhibitors treatment. In contrast, genetically encoded inhibitors did not improve HDR.
Conclusion: The ADA2 p. c.506G>A variant can be effectively edited employing surrogate strategy in HSPCs without impairing functionality. Although pharmacological inhibition of NHEJ enhances HDR efficiency, it also increases the risk of on-target chromosome aberrations, highlighting the need for careful consideration of the associated risks and benefits in therapeutic gene editing.
Pu Chen, Kim My Le, Weiwei Li, Francisca Hinrichsen, Xiaonan Liu, Ragnhild Selvig Braathen, Nanni Mamia, Monika Szymanska, Zhuokun Li, Luca Trotta, Henrikki Almusa, Katariina Mamia, Virpi Glumoff, Peter Holland, Simon Anders, Artem Kalinichenko, Meri Kaustio, Mikko R.J. Seppänen, Emma Maria Haapaniemi, Markku Varjosalo, Timi Martelius, Juha Grönholm, Janna Saarela
Background: The transmembrane adaptor protein SIT1 (signaling threshold–regulating transmembrane adaptor 1) is a negative regulator of T-cell receptor (TCR) signaling, but its role in human immunity remains poorly characterized.
Objective: We sought to investigate the molecular consequences of a biallelic loss-of-function SIT1 variant in a patient with combined immune deficiency and recurrent lymphoma.
Methods: Exome sequencing identified the putative disease-causing variant. Immunophenotyping and functional assays were performed on patient-derived lymphocytes and CRISPR-Cas9–mediated SIT1 knockout (KO) T cells from healthy donors. Transcriptomics, proteomics, and super-resolution imaging were used to characterize SIT1 deficiency.
Results: The c.236+2T>G variant caused exon 2 skipping and complete SIT1 loss. Patient/KO T cells revealed skewed T-cell subsets, increased activation and proliferation, and impaired CD8+ cytotoxicity. Transcriptomic analysis demonstrated that SIT1 KO upregulated cytokine signaling, metabolic reprogramming, and cell cycle hallmarks. Proteomic analysis confirmed SIT1’s association with Src and receptor tyrosine kinases and suggested a novel role for SIT1 in intracellular vesicle trafficking. Imaging revealed SIT1 localized to plasma membrane and TCR microclusters, colocalizing with endolysosomal compartments and linker for activation of T cells. Patient T cells exhibited impaired immune synapse maturation and vesicle accumulation on TCR stimulation. Population data link this rare SIT1 variant to lymphoid malignancy and reduced risk of autoimmunity.
Conclusions: We report the first patient with biallelic loss-of-function SIT1 variant, establishing SIT1 as a critical regulator of T-cell activation, proliferation, and synapse organization, implicating SIT1 deficiency as a novel cause of combined immunodeficiency, predisposing to lymphoid malignancy.
Katariina Mamia et al.
CRISPR-Cas9 gene editing is a promising tool to correct pathogenic variants for autologous cell therapies targeting inborn errors of immunity (IEI). Current strategies, such as gene knockout or cDNA knockin, address many single-gene defects but can disrupt gene expression, highlighting the need for precise correction platforms. While transplanting corrected autologous hematopoietic stem cells is a curative approach, it is unsuitable for patients with advanced disease, inflammation, or acute infections.
As correcting T cells is an alternative therapeutic strategy for lymphoid IEIs, we present an efficient T cell single-nucleotide variant (SNV) correction platform based on homology-directed repair (HDR). By using STAT1 gain-of-function, cartilage hair hypoplasia, deficiency of ADA2, and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy as IEI models, we demonstrate that our platform achieves up to 80% correction, with resultant functional correction of the disease phenotype in the selected models. Furthermore, we performed safety profiling using GUIDE-seq, single-cell RNA sequencing, long-read genome sequencing, and proteomics analysis and detected no genomic, transcriptomic, or proteomic aberrations.
This study establishes HDR-based SNV editing as a portable method for developing clinical autologous T cell therapies and represents a promising step toward a broad-spectrum gene correction platform for treating diverse monogenic immune disorders.
Kaustio Meri, Szymanska Monika, Li Weiwei, Braathen Ragnhild, Campbell Tessa M, Haugen Frida L, Dahal-Koirala Shiva, Silventoinen Kristiina, Nurmi Katariina, Dinius Matas, Nowlan Kirsten, Hetemäki Iivo, Chen Pu, Mamia Katariina, Seppänen Mikko, Grönholm Juha, Kekäläinen Eliisa, Haapaniemi Emma M, Aalto Kristiina, Martelius Timi, Bryceson Yenan, Eklund Kari K, Saarela Janna
Background: MAP4K1 encodes Hematopoietic Progenitor Kinase 1 (HPK1), a serine/threonine kinase that negatively regulates T cell receptor (TCR) signaling via phosphorylation of the adaptor proteins SLP-76 and Gads. While common MAP4K1 variants have been implicated in polygenic immunemediated diseases, the impact of rare germline variants on human immunity remains undefined.
Objective: To investigate the immunological and functional consequences of HPK1 deficiency in individuals with suspected Inborn Errors of Immunity (IEI).
Methods: We performed genomic linkage analysis and exome sequencing to identify disease-associated variants in IEI patients. Immunophenotyping, RNA-sequencing and functional assays were conducted on patient-derived lymphocytes, complemented by CRISPR-Cas9-mediated MAP4K1 disruption and correction in primary T cells.
Results: Heterozygous MAP4K1 loss-of-function variants were identified in two kindreds presenting with diverse immune dysregulatory symptoms, including recurrent fevers, inflammatory arthritis, EBV-related complications, and nephritis. These variants led to reduced HPK1 protein levels and SLP-76Ser376 phosphorylation. While lymphocyte development was largely preserved, T cells from HPK1-deficient individuals displayed hyperresponsiveness to TCR stimulation, characterized by elevated secretion of proinflammatory cytokines, particularly interferon-γ (IFN-γ) and tumor necrosis factor (TNF).
CRISPR-mediated knockout recapitulated, and variant-correction partially reversed this phenotype. Transcriptomic profiling of stimulated CD4⁺ T cells further revealed upregulation of immune signaling pathways—including NF-κB, JAK/STAT, and AP-1—as well as increased expression of multiple T cell cytokines, consistent with enhanced TCR signaling and T cell responses in HPK1-deficient
individuals.
Conclusion: HPK1 deficiency, caused by heterozygous loss of MAP4K1, is a novel monogenic cause of immune dysregulation. Increased T cell activation and pro-inflammatory cytokine production are implicated in disease pathogenesis.
Kornel Labun, Oline Rio, Håkon Tjeldnes, Michał Swirski, Anna Zofia Komisarczuk, Emma Haapaniemi, Eivind Valen
CRISPR/Cas is a revolutionary technology for genome editing. Although hailed as a potential cure for a wide range of genetic disorders, CRISPR/Cas translation faces severe challenges due to unintended off-target editing. Predicting these off-targets are difficult and necessitates trade-offs between speed and sensitivity.
Here, we develop the original concept of symbolic alignments to efficiently identify off-targets without sacrificing sensitivity. We also introduce data structures that allow near-instant alignment-free probabilistic ranking of guides based on their off-target counts. Implemented in the tool CHOPOFF, these innovations support mismatches, bulges and genomic sequence variation for personalized genomes while outperforming state-of-the-art methods in both speed and accuracy.
Pavel Kopcil, Carolina W. Ervik, Ganna Reint, Katariina Mamia, Monika Szymanska, Shiva Dahal-Koirala, Jacob Conradi, Sigrid Fu Skjelbostad, Oda A. Dønåsen, Xiaojun Jiang, Cecilia Fahlquist-Hagert, Oddrun Kristiansen, Trond M. Michelsen, Espen Melum, Rasmus O. Bak, Anna Komisarczuk, Emma Haapaniemi
Background: The homozygous ADA2: c.506G>A (p.Arg169Gln; p.R169Q) variant accounts for majority of Deficiency in Adenosine Deaminase 2 (DADA2). This monogenic disorder may be amenable to ex vivo gene therapy by correcting the pathogenic mutation in CD34+ hematopoietic stem and progenitor cells (HSPCs).
Objective: To apply CRISPR-Cas9 and homology-directed repair (HDR) as a surrogate strategy to model correction of the pathogenic ADA2 c.506G>A variant in healthy cord blood HSPCs.
Methods: HSPCs were electroporated with optimised CRISPR-Cas9 editing reagents, and editing outcomes, including HDR and on-target deletions, were quantified by ddPCR. Cell functionality was assessed through colony-forming unit (CFU) assays and by xenotransplantation into NOD SCID Gamma (NSG) mice. Two HDR enhancement strategies were tested: (1) genetic inhibitors of p53 and non-homologous end joining (NHEJ) pathways, and (2) pharmacological NHEJ inhibition.
Results: Small-molecule NHEJ inhibitors increased HDR efficiency approximately two-fold (from ∼40 % to ∼80 %). Edited HSPCs retained normal CFU capacity and successfully engrafted in NSG mice. However, up to 8 % of edited cells exhibited on-target chromosome loss, though this declined over time. Up to 40 % of T cells and fibroblasts demonstrated similar losses under NHEJ inhibitors treatment. In contrast, genetically encoded inhibitors did not improve HDR.
Conclusion: The ADA2 p. c.506G>A variant can be effectively edited employing surrogate strategy in HSPCs without impairing functionality. Although pharmacological inhibition of NHEJ enhances HDR efficiency, it also increases the risk of on-target chromosome aberrations, highlighting the need for careful consideration of the associated risks and benefits in therapeutic gene editing.
Inga S Sakovich, Emma Haapaniemi; PID_team_Belarus; Janna Saarela, Svetlana O Sharapova. J Clin Immunol. 2023 Dec 22 44(1):9.
No abstract available.
Zhuokun Li, Ganna Reint, Emma Haapaniemi
Summary
CRISPR-Cas9 gene editing is an efficient technique to modify specific sites/regions of DNA. Delivery of the Cas9 by mRNA is particularly promising in pre-clinical genome editing applications for its transient, nonintegrating feature.
However, the off-target of Cas9-gRNA still remains a concern and needs a specific monitor. Here, we present a revised protocol to edit fibroblasts by in vitro transcribed Cas9 mRNA and profile its off-target effect by the optimized GUIDE-seq method. This protocol can also be applied to other cell lines.
Leiding JW et al.
Background
In 2014, germline STAT3 gain of function (GOF) mutations were first described to cause a novel multi-system disease of early onset lymphoproliferation and autoimmunity.
Objective
This pivotal cohort study defines the scope, natural history, treatment, and overall survival of a large global cohort of patients with pathogenic STAT3 GOF variants.
Methods
We identified 191 patients from 33 countries with 72 unique mutations. Inclusion criteria included symptoms of immune dysregulation and a biochemically confirmed germline heterozygous GOF variant in STAT3.
Results
Overall survival was 88%, median age at onset of symptoms was 2.3 years and median age at diagnosis was 12 years. Immune dysregulatory features were present in all patients: lymphoproliferation was the most common manifestation (73%); increased frequencies of double negative (CD4-CD8-) T cells were found in 83% of patients tested. Autoimmune cytopenias were the second most common clinical manifestation (67%), followed by growth delay, enteropathy, skin disease, pulmonary disease, endocrinopathy, arthritis, autoimmune hepatitis, neurologic disease, vasculopathy, renal disease and malignancy.
Infections were reported in 72% of the cohort. A cellular and humoral immunodeficiency was observed in 37% and 51% of patients, respectively. Clinical symptoms dramatically improved in patients treated with JAK inhibitors while a variety of other immunomodulatory treatment modalities were less efficacious. Thus far, 23 patients have undergone bone marrow transplant with 62% survival rate.
Conclusion
STAT3 GOF patients present with a wide array of immune-mediated disease including lymphoproliferation, autoimmune cytopenias and multisystem autoimmunity. Patient care tends to be siloed without a clear treatment strategy. Thus, early identification and prompt treatment implementation are lifesaving for STAT3 GOF syndrome.
Ganna Reint*, Zhuokun Li*, Kornel Labun, Salla Keskitalo, Inkeri Soppa, Katariina Mamia, Eero Tolo, Monika Szymanska, Leonardo A Meza-Zepeda, Susanne Lorenz, Artur Cieslar-Pobuda, Xian Hu1, Diana L Bordin, Judith Staerk, Eivind Valen, Bernhard Schmierer, Markku Varjosalo, Jussi Taipale, Emma Haapaniemi, eLife 2021;10:e75415
Precision CRISPR gene editing relies on the cellular homology-directed DNA repair (HDR) to introduce custom DNA sequences to target sites. The HDR editing efficiency varies between cell types and genomic sites, and the sources of this variation are incompletely understood.
Here, we have studied the effect of 450 DNA repair protein-Cas9 fusions on CRISPR genome editing outcomes. We find the majority of fusions to improve precision genome editing only modestly in a locus- and cell-type specific manner. We identify Cas9-POLD3 fusion that enhances editing by speeding up the initiation of DNA repair.
We conclude that while DNA repair protein fusions to Cas9 can improve HDR CRISPR editing, most need to be optimized to the cell type and genomic site, highlighting the diversity of factors contributing to locus-specific genome editing outcomes.
Kaustio M, Nayebzadeh N, Hinttala R, Tapiainen T, Åström P, Mamia K, Pernaa N, Lehtonen J, Glumoff V, Rahikkala E, Honkila M, Olsén P, Hassinen A, Polso M, Al Sukaiti N, Al Shekaili J, Al Kindi M, Al Hashmi N, Almusa H, Bulanova D, Haapaniemi E, Chen P, Suo-Palosaari M, Vieira P, Tuominen H, Kokkonen H, Al Macki N, Al Habsi H, Löppönen T, Rantala H, Pietiäinen V, Zhang SY, Renko M, Hautala T, Al Farsi T, Uusimaa J, Saarela J.
Background:
Homozygous loss of DIAPH1 results in seizures, cortical blindness, and microcephaly syndrome (SCBMS). We studied 5 Finnish and 2 Omani patients with loss of DIAPH1 presenting with SCBMS, mitochondrial dysfunction, and immunodeficiency.
Objective:
We sought to further characterize phenotypes and disease mechanisms associated with loss of DIAPH1.
Methods:
Exome sequencing, genotyping and haplotype analysis, B- and T-cell phenotyping, in vitro lymphocyte stimulation assays, analyses of mitochondrial function, immunofluorescence staining for cytoskeletal proteins and mitochondria, and CRISPR-Cas9 DIAPH1 knockout in heathy donor PBMCs were used.
Results:
Genetic analyses found all Finnish patients homozygous for a rare DIAPH1 splice-variant (NM_005219:c.684+1G>A) enriched in the Finnish population, and Omani patients homozygous for a previously described pathogenic DIAPH1 frameshift-variant (NM_005219:c.2769delT;p.F923fs).
In addition to microcephaly, epilepsy, and cortical blindness characteristic to SCBMS, the patients presented with infection susceptibility due to defective lymphocyte maturation and 3 patients developed B-cell lymphoma. Patients’ immunophenotype was characterized by poor lymphocyte activation and proliferation, defective B-cell maturation, and lack of naive T cells. CRISPR-Cas9 knockout of DIAPH1 in PBMCs from healthy donors replicated the T-cell activation defect. Patient-derived peripheral blood T cells exhibited impaired adhesion and inefficient microtubule-organizing center repositioning to the immunologic synapse. The clinical symptoms and laboratory tests also suggested mitochondrial dysfunction. Experiments with immortalized, patient-derived fibroblasts indicated that DIAPH1 affects the amount of complex IV of the mitochondrial respiratory chain.
Conclusions:
Our data demonstrate that individuals with SCBMS can have combined immune deficiency and implicate defective cytoskeletal organization and mitochondrial dysfunction in SCBMS pathogenesis.
Keskitalo S, Haapaniemi E, Einarsdottir E, Rajamäki K, Heikkilä H, Ilander M, Pöyhönen M, Morgunova E, Hokynar K, Lagström S, Kivirikko S, Mustjoki S, Eklund K, Saarela J, Kere J, Seppänen MRJ, Ranki A, Hannula-Jouppi K, Varjosalo M.
Upon binding to pathogen or self-derived cytosolic nucleic acids cyclic GMP-AMP synthase (cGAS) triggers the production of cGAMP that further activates transmembrane protein STING. Upon activation STING translocates from ER via Golgi to vesicles.
Monogenic STING gain-of-function mutations cause early-onset type I interferonopathy, with disease presentation ranging from fatal vasculopathy to mild chilblain lupus. Molecular mechanisms underlying the variable phenotype-genotype correlation are presently unclear.
Here, we report a novel gain-of-function G207E STING mutation causing a distinct phenotype with alopecia, photosensitivity, thyroid dysfunction, and features of STING-associated vasculopathy with onset in infancy (SAVI), such as livedo reticularis, skin vasculitis, nasal septum perforation, facial erythema, and bacterial infections.
Polymorphism in TMEM173 and IFIH1 showed variable penetrance in the affected family, implying contribution to varying phenotype spectrum. The G207E mutation constitutively activates inflammation-related pathways in vitro, and causes aberrant interferon signature and inflammasome activation in patient PBMCs.
Treatment with Janus kinase 1 and 2 (JAK1/2) inhibitor baricitinib was beneficiary for a vasculitic ulcer, induced hair regrowth and improved overall well-being in one patient. Protein-protein interactions propose impaired cellular trafficking of G207E mutant. These findings reveal the molecular landscape of STING and propose common polymorphisms in TMEM173 and IFIH1 as likely modifiers of the phenotype.
Emma Haapaniemi, Sandeep Botla, Jenna Persson, Bernhard Schmierer, Jussi Taipale. Mol Syst Biol. 2019 Aug 15(8)
Haapaniemi et al address the issues raised by Brown et al and discuss several differences between the analyses performed by the two groups.
Sharapova SO, Haapaniemi E, Sakovich IS, Kostyuchenko LV, Donkó A, Dulau-Florea A, Malko O, Bondarenko AV, Stegantseva MV, Leto TL, Uygun V, Karasu GT, Holland SM, Hsu AP, Aleinikova OV. J. Clin Immunol. 2019 May 6 [Epub ahead of print].
Here we describe a 10-year-old girl with combined immunodeficiency presenting as recurring chest infections, lung disease and herpetic skin infections. The patient experienced two hematopoietic stem cell transplantations and despite full chimerism, she developed bone marrow aplasia due to adenovirus infection and died at post-transplant day 86.
Immunologic investigation revealed low numbers of TRECs/KRECs, a severe reduction of memory B cells, absence of isohemagglutinins, and low IgG levels. Whole exome sequencing (WES) identified a novel heterozygous mutation in RAC2(c.275A > C, p.N92 T). Flow cytometric investigation of neutrophil migration demonstrated an absence of chemotaxis to fMLP. Cell lines transfected with RAC2 [N92 T] displayed characteristics of active GTP-bound RAC2 including enhanced reactive oxygen species (ROS) production both at rest and in response to PMA. Our findings broaden the clinical picture of RAC2 dysfunction, showing that some individuals can present with a combined immunodeficiency later in childhood rather than a congenital neutrophil disease.
Keskitalo S*, Haapaniemi E*, Glumoff V, Liu X, Fogarty C, Lehtinen V, Rajala H, Chian-Cher S, Mustjoki S, Kovanen P, Lohi J, Bryceson Y, Seppänen M, Kere J, Heiskanen K, Varjosalo M. Genomic Medicine 4, 14 (2019)
Mutations in several proteins functioning as endolysosomal components cause monogenic autoimmune diseases, of which pathogenesis is linked to increased endoplasmic reticulum stress, inefficient autophagy, and defective recycling of immune receptors.
We report here a heterozygous TOM1 p.G307D missense mutation, detected by whole-exome sequencing, in two related patients presenting with early-onset autoimmunity, antibody deficiency, and features of combined immunodeficiency.
The index patient suffered from recurrent respiratory tract infections and oligoarthritis since early teens, and later developed persistent low-copy EBV-viremia, as well as an antibody deficiency. Her infant son developed hypogammaglobulinemia, autoimmune enteropathy, interstitial lung disease, profound growth failure, and treatment-resistant psoriasis vulgaris.
Consistent with previous knowledge on TOM1 protein function, we detected impaired autophagy and enhanced susceptibility to apoptosis in patient-derived cells. In addition, we noted diminished STAT and ERK1/2 signaling in patient fibroblasts, as well as poor IFN-γ and IL-17 secretion in T cells. The mutant TOM1 failed to interact with TOLLIP, a protein required for IL-1 recycling, PAMP signaling and autophagosome maturation, further strengthening the link between the candidate mutation and patient pathophysiology.
In sum, we report here an identification of a novel gene, TOM1, associating with early-onset autoimmunity, antibody deficiency, and features of combined immunodeficiency. Other patient cases from unrelated families are needed to firmly establish a causal relationship between the genotype and the phenotype.
Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. Nat Med, 2018. 24(7): p. 927-930
In this publication we report that genome editing by CRISPR–Cas9 induces a p53-mediated DNA damage response and cell cycle arrest in immortalized human retinal pigment epithelial cells, leading to a selection against cells with a functional p53 pathway. Inhibition of p53 prevents the damage response and increases the rate of homologous recombination from a donor template.
These results suggest that p53 inhibition may improve the efficiency of genome editing of untransformed cells and that p53 function should be monitored when developing cell-based therapies utilizing CRISPR–Cas9.
Sharapova S, Haapaniemi E, Sakovich I, Rojas J, Gámez-Díaz L, Mareika Y, Guryanova I, Migas A, Mikhaleuskaya T, Grimbacher B, Aleinikova O. J Clin Immunol. 2018 38(4): p.471-474
Here, we report the clinical, genetic, and immunological data from two siblings in a Belarusian family with a homozygous, truncating LRBA mutation (c.2762G>C, p.Ser921Stop). Patient 1 (P1), male, was born at term to non-consanguineous parents.
Kaustio M*, Haapaniemi E*, Nurkkala H*, Helminen M, Park G, Syrjänen J, Einarsdottir E, Sahu B, Kilpinen S, Rounioja S, Fogarty C, Glumoff V, Kulmala P, Katayama S, Tamene F, Trotta L, Morungova E, Krjutskov K, Martelius T, Mustjoki S, Taipale J, Saarela J, Kere J, Varjosalo M, Seppänen M. Journal of Allergy and Clinical Immunology 2017. 40(3): p.782-796
Background
The nuclear factor κ light-chain enhancer of activated B cells (NF-κB) signaling pathway is a key regulator of immune responses. Accordingly, mutations in several NF-κB pathway genes cause immunodeficiency.
Objective
We sought to identify the cause of disease in 3 unrelated Finnish kindreds with variable symptoms of immunodeficiency and autoinflammation.
Methods
We applied genetic linkage analysis and next-generation sequencing and functional analyses of NFKB1 and its mutated alleles.
Results
In all affected subjects we detected novel heterozygous variants in NFKB1, encoding for p50/p105. Symptoms in variant carriers differed depending on the mutation. Patients harboring a p.I553M variant presented with antibody deficiency, infection susceptibility, and multiorgan autoimmunity. Patients with a p.H67R substitution had antibody deficiency and experienced autoinflammatory episodes, including aphthae, gastrointestinal disease, febrile attacks, and small-vessel vasculitis characteristic of Behçet disease. Patients with a p.R157X stop-gain experienced hyperinflammatory responses to surgery and showed enhanced inflammasome activation. In functional analyses the p.R157X variant caused proteasome-dependent degradation of both the truncated and wild-type proteins, leading to a dramatic loss of p50/p105. The p.H67R variant reduced nuclear entry of p50 and showed decreased transcriptional activity in luciferase reporter assays. The p.I553M mutation in turn showed no change in p50 function but exhibited reduced p105 phosphorylation and stability. Affinity purification mass spectrometry also demonstrated that both missense variants led to altered protein-protein interactions.
Conclusion
Our findings broaden the scope of phenotypes caused by mutations in NFKB1 and suggest that a subset of autoinflammatory diseases, such as Behçet disease, can be caused by rare monogenic variants in genes of the NF-κB pathway.
Haapaniemi E, Fogarty C, Katayama S, Vihinen H, Keskitalo S, Ilander M, Krjutškov K, Mustjoki S, Lehto M, Hautala T, Jokitalo E, Velagapudi V, Varjosalo M, Seppänen M and Kere J. Journal of Allergy and Clinical Immunology 2017 Apr;139(4): p.1391-1393
No abstract available.
Dobbs K, Dominguez‐Conde C Zhang SY, Parolini S, Audry M, Chou J, Haapaniemi E, Keles S, Bilic I, Okada S, Massaad MJ, Rounioja S, Alwahadneh AM, Serwas NK, Capuder K, Ciftci E, Felgentreff K, Ohsumi T, Pedergnana V, Boisson B, Haskoloğlu S, Ensari A, Schuster M, Moretta A, Itan Y, Patrizi O, Rozenberg F, Lebon P, Saarela J, Knip M, Petrovski S, Goldstein DB, Parrott RE, Savas B, Schambach A, Tabellini G, Bock C, Chatila T, Comeau AM, Geha RS, Abel L, Buckley RH, Ikincioğullari A, Al‐Herz W, Helminen M, Doğu F, Casanova JL, Boztuğ K, Notarangelo LD. N Engl J Med 2015; 372:2409-2422
Background
Combined immunodeficiencies are marked by inborn errors of T-cell immunity in which the T cells that are present are quantitatively or functionally deficient. Impaired humoral immunity is also common. Patients have severe infections, autoimmunity, or both. The specific molecular, cellular, and clinical features of many types of combined immunodeficiencies remain unknown.
Methods
We performed genetic and cellular immunologic studies involving five unrelated children with early-onset invasive bacterial and viral infections, lymphopenia, and defective T-cell, B-cell, and natural killer (NK)–cell responses. Two patients died early in childhood; after allogeneic hematopoietic stem-cell transplantation, the other three had normalization of T-cell function and clinical improvement.
Results
We identified biallelic mutations in the dedicator of cytokinesis 2 gene (DOCK2) in these five patients. RAC1 activation was impaired in the T cells. Chemokine-induced migration and actin polymerization were defective in the T cells, B cells, and NK cells. NK-cell degranulation was also affected. Interferon-α and interferon-λ production by peripheral-blood mononuclear cells was diminished after viral infection. Moreover, in DOCK2-deficient fibroblasts, viral replication was increased and virus-induced cell death was enhanced; these conditions were normalized by treatment with interferon alfa-2b or after expression of wild-type DOCK2.
Conclusions
Autosomal recessive DOCK2 deficiency is a new mendelian disorder with pleiotropic defects of hematopoietic and nonhematopoietic immunity. Children with clinical features of combined immunodeficiencies, especially with early-onset, invasive infections, may have this condition. (Supported by the National Institutes of Health and others.)
Haapaniemi E, Kaustio M, Rajala H, van Adrichem A, Kainulainen L, Glumoff V, Doffinger R, Kuusanmäki H, Heiskanen-Kosma T, Trotta L, Chiang S, Kulmala P, Eldfors S, Katainen R, Siitonen S, Karjalainen-Lindsberg M, Kovanen P, Otonkoski T, Porkka K, Heiskanen K, Hänninen A, Bryceson Y, Uusitalo-Seppälä R, Saarela J, Seppänen M, Mustjoki S, Kere J. Blood, 2015. 125(4): p. 639-48.
The signal transducer and activator of transcription (STAT) family of transcription factors orchestrate hematopoietic cell differentiation. Recently, mutations in STAT1, STAT5B, and STAT3 have been linked to development of immunodysregulation polyendocrinopathy enteropathy X-linked–like syndrome.
Here, we immunologically characterized 3 patients with de novo activating mutations in the DNA binding or dimerization domains of STAT3 (p.K392R, p.M394T, and p.K658N, respectively). The patients displayed multiorgan autoimmunity, lymphoproliferation, and delayed-onset mycobacterial disease. Immunologically, we noted hypogammaglobulinemia with terminal B-cell maturation arrest, dendritic cell deficiency, peripheral eosinopenia, increased double-negative (CD4−CD8−) T cells, and decreased natural killer, T helper 17, and regulatory T-cell numbers. Notably, the patient harboring the K392R mutation developed T-cell large granular lymphocytic leukemia at age 14 years.
Our results broaden the spectrum of phenotypes caused by activating STAT3 mutations, highlight the role of STAT3 in the development and differentiation of multiple immune cell lineages, and strengthen the link between the STAT family of transcription factors and autoimmunity.
Flanagan S.E*, Haapaniemi E*, Russel M*, Caswell R, Lango Allen H, De Franco E, McDonald T, Rajala H, Ramelius A, Barton J, Heiskanen K, Heiskanen-Kosma T, Kajosaari M, Murphy N, Milenkovic T, Seppänen M, Lenmark Å, Mustjoki S, Otonkoski T, Kere J, Morgan N, Ellard S, Hattersley A. Nat Genet, 2014. 46(8): p. 812-4
Monogenic causes of autoimmunity provide key insights into the complex regulation of the immune system. We report a new monogenic cause of autoimmunity resulting from de novo germline activating STAT3 mutations in five individuals with a spectrum of early-onset autoimmune disease, including type 1 diabetes. These findings emphasize the critical role of STAT3 in autoimmune disease and contrast with the germline inactivating STAT3 mutations that result in hyper IgE syndrome.
* Equal contribution
{kind=link}
{kind=link}